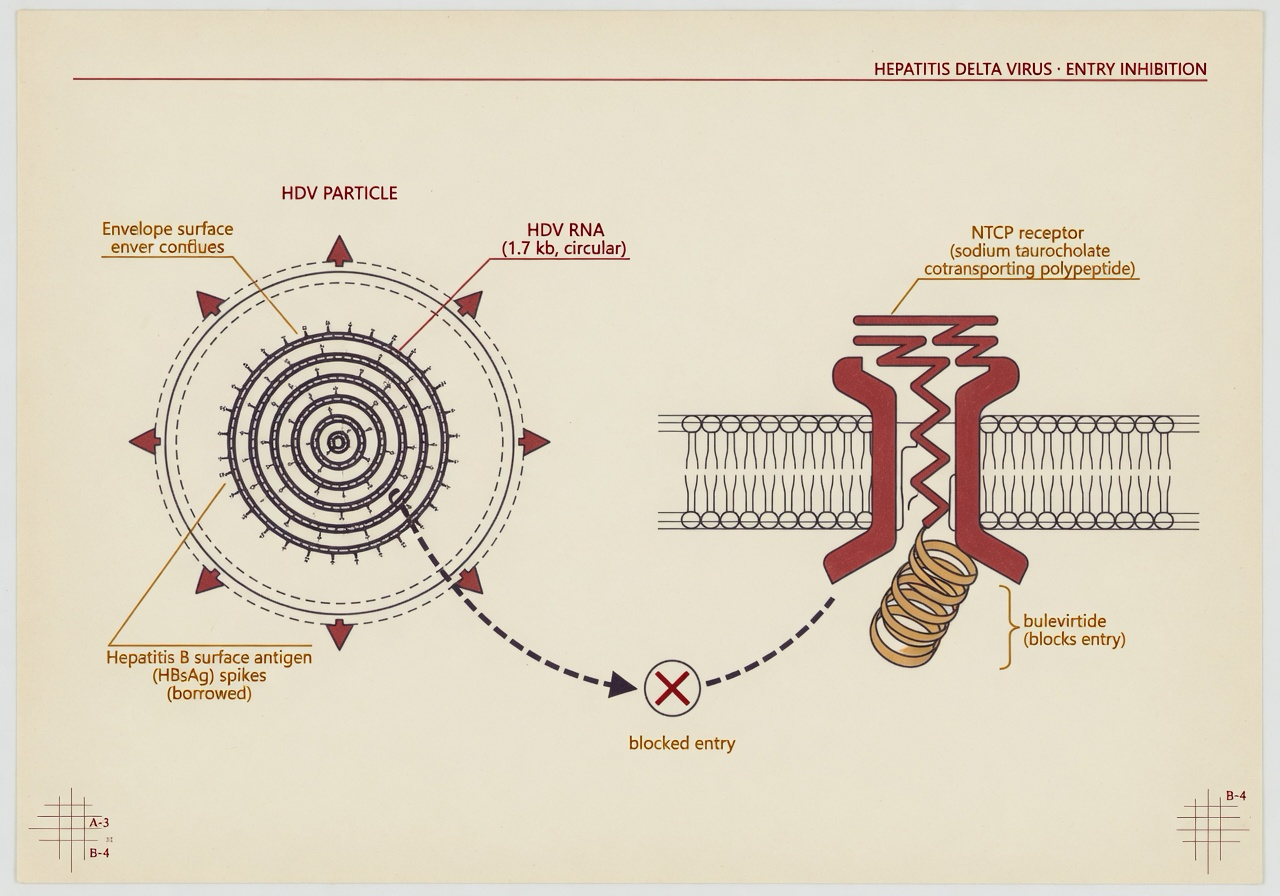
The U.S. Food and Drug Administration on May 22, 2026 granted accelerated approval to Hepcludex (bulevirtide-gmod), an 8.5 mg once-daily subcutaneous injection from Gilead Sciences, for the treatment of chronic hepatitis delta virus (HDV) infection in adults without cirrhosis or with compensated cirrhosis. It is the first and only HDV antiviral cleared for use in the United States. The agency’s approval is based on reductions in HDV RNA and normalization of alanine aminotransferase observed at Week 48 of the Phase 3 MYR301 trial; continued approval is contingent on a confirmatory long-term outcomes study, which Gilead says is already underway. The U.S. approval comes nearly six years after the European Medicines Agency cleared the same drug in July 2020.
What was approved, and on what evidence
The approved label, per Gilead’s May 22 announcement, reads: “Hepcludex (bulevirtide-gmod) 8.5 mg for injection is indicated for the treatment of chronic hepatitis delta virus infection in adults without cirrhosis or with compensated cirrhosis.” The label also carries an explicit caveat that an improvement in disease-related clinical outcomes has not been established and that the decision rests on intermediate endpoints. The MYR301 trial (NCT03852719), published in the New England Journal of Medicine in 2023, randomized 150 adults with chronic HDV and compensated liver disease in a 1:1:1 ratio to no treatment, bulevirtide 2 mg per day, or bulevirtide 10 mg per day for 48 weeks. The primary endpoint was a combined response: undetectable HDV RNA or a fall of at least two log10 IU per milliliter from baseline, together with ALT normalization. At Week 48, approximately 45 percent of the 2 mg arm and 48 percent of the 10 mg arm met the combined response, against 2 percent in the no-treatment arm.
The dose that the FDA approved, 8.5 mg subcutaneous once daily, is higher than the 2 mg dose authorized by the EMA on the same trial. Gilead has not publicly explained the dose selection in detail; the company’s May 22 release notes that the U.S. label is based on the MYR301 data and its open-label extension. The Phase 3 study did not establish that one dose was superior to the other on the primary virologic-biochemical endpoint, and the FDA’s acceptance of the higher dose appears to be driven by post-baseline exposure-response considerations rather than head-to-head superiority.
Accelerated approval, and what it commits Gilead to
The pathway matters. Accelerated approval permits the FDA to clear a therapy on the basis of an intermediate endpoint reasonably likely to predict clinical benefit, in exchange for a sponsor commitment to verify benefit through a confirmatory trial. Hepcludex was previously granted Breakthrough Therapy and Orphan Drug designations, both of which speak to development priority rather than to the strength of the evidence on which approval rests. The confirmatory study is a long-term outcomes trial Gilead has already initiated; the company has not published the protocol or the prespecified clinical endpoint, but for HDV the candidates are well established: progression to cirrhosis, decompensation events, hepatocellular carcinoma, liver transplant, and death. None of those endpoints accrue at 48 weeks. The confirmatory window will therefore run on a different timescale than the trial that earned the approval. If the confirmatory trial fails to verify clinical benefit, the FDA can withdraw the indication.
The disease the drug treats
HDV is a defective satellite virus. It cannot replicate on its own and depends on the envelope protein of hepatitis B virus (HBsAg) to assemble infectious particles, which is why HDV is found only in people already carrying HBV. The World Health Organization estimates that 12 million people worldwide, around 5 percent of those living with chronic HBV, are co-infected with HDV; the U.S. figure is not formally tracked because HDV is not a nationally notifiable condition, and the Centers for Disease Control and Prevention’s page on hepatitis D states explicitly that “the actual number of hepatitis D cases in the US is unknown.” The clinical pattern is the reason for the drug’s priority. WHO calls HDV “the most severe form of chronic viral hepatitis due to more rapid progression towards hepatocellular carcinoma and liver-related death,” and notes that superinfection of an HBV carrier with HDV accelerates progression to cirrhosis in 70 to 90 percent of cases.
Before May 22, U.S. clinicians treating chronic HDV had no on-label antiviral. Off-label pegylated interferon alfa was the only option, with sustained response in roughly 20 to 30 percent of treated patients and significant tolerability problems. The clinical gap was therefore not in dispute. What was in dispute was when, and on what U.S. evidence package, an HDV antiviral would clear the FDA.
An improvement in disease-related clinical outcomes has not been established. — FDA-approved label for Hepcludex (bulevirtide-gmod), as reproduced in Gilead’s May 22, 2026 announcement
Why the U.S. came late
The EMA granted conditional marketing authorization for Hepcludex on July 31, 2020, and converted it to standard authorization on July 18, 2023. Gilead first filed a U.S. biologics license application in 2021. The FDA issued a Complete Response Letter on the application; reporting at the time, and Gilead’s subsequent disclosures, indicated that the response cited manufacturing-related deficiencies rather than questions about the clinical data. The May 22 approval follows a resubmission addressing those issues. The six-year gap between EU and U.S. authorization is therefore not a story about clinical doubt, and the FDA has not pointed to the MYR301 data as the limiting factor. It is a story about how a small-molecule peptide injectable, with a small commercial footprint and a complex chemistry-manufacturing-and-controls package, moved through the U.S. system. The drug being approved at Week 48 of the same trial the EMA used almost six years earlier underscores that distinction.
What remains uncertain
Three uncertainties deserve to be stated cleanly. First, durability. MYR301 measured the combined response at 48 weeks and through the open-label extension; the field has no established sustained virologic response endpoint for HDV analogous to the SVR12 used for hepatitis C. Whether on-treatment suppression translates into off-treatment cure, or whether bulevirtide is effectively a chronic suppressive therapy, is the question the confirmatory trial is designed to address.
Second, the clinical endpoint. Virologic suppression and ALT normalization are reasonable surrogates; they are not the outcomes patients live and die by. Until the confirmatory long-term outcomes study reports, the central evidentiary claim of this approval, that a 48-week intermediate response will translate into reduced cirrhosis, fewer hepatocellular carcinomas, and lower liver-related mortality, remains a hypothesis the FDA has judged reasonably likely.
Third, access. Gilead has not disclosed a U.S. list price for Hepcludex. The company has pointed to its existing Support Path program for coverage assistance; what the launch price will be, how payers will cover a daily subcutaneous injectable for a small and underdiagnosed patient population, and how diagnosis itself will scale up given that HDV is not a nationally notifiable condition, are open questions the May 22 release does not answer. The drug is approved; the system that delivers it to patients with chronic HDV is, as of this week, still being assembled.
What to track
Three specifics on the calendar. The first is the confirmatory long-term outcomes trial. Gilead has confirmed it is underway; the protocol, the prespecified primary clinical endpoint, and the projected readout window will be the substantive checkpoint on whether accelerated approval becomes regular approval. The second is the prescribing information itself. The label posted to Drugs@FDA, and the labeling negotiations behind it, will indicate the boxed warning text, the specific monitoring requirements, the discontinuation language for HDV and HBV flare, and the dosing instructions for adults with compensated cirrhosis. Third is the U.S. epidemiology. Because HDV is not a notifiable condition in the United States, the denominator for the patient population this approval applies to is, today, an estimate built on small seroprevalence studies. Whether HDV testing scales up in the wake of an available therapy will determine how many of the people who should be on this drug will actually be identified.